1. 서 론
이산화탄소(CO2), 메탄(CH4), 아산화질소(N2O)와 같은 온실가스에 의한 지구 온난화는 기후변화를 야기하는 것으로 잘 알려져 있다. 특히, 대기 중 CO2 농도의 증가와 기후 변화는 밀접한 상관관계를 보이고 있으나, 산업화 이후 CO2 배출량은 증가세가 꺾이지 않고 있다. 이에 지난 수년간 배출되는 CO2를 포집하여 공기 중의 CO2 농도를 감축하려는 많은 방법이 연구되어왔다[1]. 다량의 CO2가 대기 중으로 배출되는 것을 방지하기 위해 오염원에서 CO2 를 포집하여 저장 위치로 옮겨 저장하는 전 과정을 탄소의 포집과 저장(carbon capture and storage, CCS)이라고 한다[2-4]. CCS 과정 중 CO2 포집은 연소 후(post combustion), 연소 전(pre combustion), 순산소 연소(oxy-fuel combustion)로 구분할 수 있다. 배기가스 중 CO2를 제거하는 기술인 연소 후 포집 기술에는 화학 흡수법이 많이 사용된다. 주로 아민 또는 암모니아 계열의 흡수제를 이용하는 화학 흡수법은 높은 효율과 공정 도입의 용이성으로 주목 받아왔다[5,6]. 특히, 아민 계열 흡수제는 화학 흡수법에 사용되는 대표적인 물질로 발전소 배기가스와 같이 CO2 농도가 5~15% 수준인 저 농도 가스처리에 적합하기에 이와 관련한 연구가 활발하게 진행되고 있다.
모노에탄올아민(monoethanolamine, MEA), 디에탄올아민(diethanolamine)과 같은 1, 2차 알카놀아민(alkanolamine) 수용액은 CO2와 반응속도가 빠르고 비용이 저렴하다는 이유로 아민 계열 흡수제 중 흔히 사용되고 있다. 이들 알카놀아민은 CO2와 반응하여 카바메이트(carbamate)를 생성하는 것으로 알려져 있다. 하지만 알카놀아민 수용액을 이용하여 CO2를 흡수하는 과정에 관한 많은 연구에도 불구하고, 그 반응 메커니즘은 아직 분명히 밝혀지지 않았다. 다음은 실험 및 이론적 연구 결과로 주로 제시되는 몇 가지 반응 메커니즘이다. (1), (2)는 카바민산(carbamic acid)이 중간체(intermediate)로 생성된 후 카바메이트가 생성되는 2단계 반응 메커니즘이다[6,7].
반응식에서 알카놀아민 분자에서 아미노 그룹의 질소에 결합한 알코올을 R로 표시했고, 수소 이온을 빼앗는 염기[물, 아민, 수산화이온(OH-)]를 B로 표시했다. 반면, (3), (4)는 위반응 메커니즘과 거의 유사하지만, 반응 중간체로 중성분자 내에 전기적으로 양성과 음성을 모두 가지는 양쪽성 이온(zwitterion)이 생성되는 2단계 반응 메커니즘이다[8-10]. (1)의 식에서 알카놀아민의 질소에 H 원자 1개와 COOH의 C가 결합한 구조를 가진 분자를 카바민산, (3)의 식에서 알카놀아민의 질소에 H 원자 2개와 COO의 C가 결합한 구조를 가진 분자를 양쪽성 이온으로 구분하였다. 알카놀아민과 이산화탄소의 반응에 대한 계산화학적 연구에서 (1), (2)와 (3), (4)의 반응 메커니즘은 구조와 에너지적인 분석에 있어 차이가 있기 때문에 이를 구분하여 서술하였다.
하지만 (5)에서도 세 개의 분자가 한꺼번에 반응한다기보다는 CO2와 알카놀아민이 반응하여 중간체를 생성하지만, 중간체의 수명(life time)이 너무 짧아 카바메이트 이온을 생성하는 염기와의 반응 과정을 따로 분리하기 어려운 것으로 볼 수도 있다.
이전의 몇몇 계산화학적 연구 결과를 통해 위에서 제시된 세가지 반응 메커니즘[(1)과 (2), (3)과 (4), (5)] 중에서 또 다른 아민 분자가 염기로 작용하여 (5)와 같은 한 단계 반응 메커니즘으로 반응한다고 보는 것이 활성화 에너지 측면에서 가장 가능성이 높은 메커니즘으로 볼 수 있다는 것이 보고되기도 했다[11,13,14]. 어떤 반응 메커니즘에 해당하든지 수소 원자 전달체의 역할이 중요했고, 이 역할에 물 분자보다는 아민 분자를 적용했을 때 활성화 에너지가 훨씬 낮다는 것을 알 수 있었다. 당연한 문제일 수 있지만, 실제 반응에서는 반응부 주변에 많은 분자들이 존재할 것이며 이들이 반응에 영향을 줄 수 있다. 하지만 계산화학적 연구에서 많은 분자들을 모두 계산 대상으로 하는 것은 계산 시간과 비용 측면에서 효율성이 떨어질 수 있어서 적절한 범위의 모델 설계가 필요하다. 본 연구에서는 반응에 참여하는 분자들 이외에 반응부 주변을 둘러싼 분자들의 영향을 추가로 고려하여 설계한 모델로 MEA 수용액에서 CO2 흡수 반응을 분석함으로써 이와 같은 반응을 이론적으로 연구하는데 주변 분자들의 영향을 어느 정도로 고려해야 할 것인지 제안하고자 한다. 이를 위해 본 논문에서는 이산화탄소가 MEA에 흡수되는 반응에 대한 저자의 이전 연구[14]와 반응부 주변에 분자들을 추가한 결과를 비교 분석할 것이다.
2. 연구방법
2.1. 계산방법
밀도범함수 이론(density functional theory, DFT)은 Hohenberg, Kohn과 Sham에 의해 제시된 분자 계산의 방법론으로 슈뢰딩거의 개별 전자 파동함수를 전자들의 밀도로 대체시켜서 원자, 분자뿐만 아니라 고체 시스템에 대한 분자 모델링(molecular modeling)을 가능하게 하였다[15-17]. 양자화학과 컴퓨팅 능력의 급격한 발달은 DFT를 이용하여 반응물, 생성물, 반응 중간체의 분자구조와 진동 운동 주파수(vibrational frequency), 에너지, 반응 메커니즘 등을 비교적 정확하고 빠르게 계산할 수 있게 하여 실험을 통해 얻은 결과를 검증하는 것과 더불어 이를 활용한 독자적인 이론적 연구도 가능하게 하였다[18-20].
MEA 수용액에 CO2가 흡수되는 반응 메커니즘을 분석하기 위한 모든 계산은 Gaussian 09 프로그램을 사용하여 수행하였다[21]. DFT를 이용하여 반응물, 생성물, 중간체, 전이 상태(transition state)의 구조와 에너지, 진동수(frequency) 등을 계산하였다. 모든 구조는 B3LYP(Becke's three parameterized Lee-Yand-Parr exchange) [22] 혼성 함수(hybrid functional)를 이용했고, 기저 집합(basis set)은 6-311G+(d, p)를 사용했으며, 수용액에서의 반응을 연구하기 위해 PCM(polarizable continuum model)을 적용했다.
구조 최적화 계산으로 얻은 반응물, 생성물, 중간체(intermediate) 구조는 모두 진동수 계산을 통해 에너지적으로 지역 최저값(local minima)에 해당하는 구조(모두 양의 진동수)라는 것을 확인하였고, 전이 상태 구조의 한 개의 음의 진동수가 반응 좌표(reaction coordinate)를 보여주는 것을 통해 안장점(saddle point)에 해당하는 구조임을 확인했다. 또한, IRC(intrinsic reaction coordinate) 계산을 통해 얻어진 반응물과 생성물의 구조를 확인함으로써 계산된 전이 상태가 CO2 흡수 반응을 제대로 나타내는 것인지 검증하였다. 활성화 에너지를 기준으로 반응이 일어날 가능성이 높은 반응 메커니즘을 제안하는 연구라서 제대로 된 전이상태의 구조를 찾는 일이 이 연구에서 무엇보다 중요했다.
2.2. 계산 모델
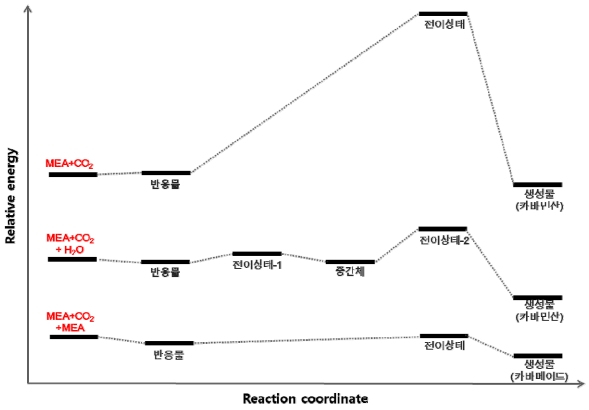
Fig. 1은 저자의 이전 연구[14]에서 아민 수용액에 CO2가 흡수되는 반응을 세가지 반응 모델로 구분하여 분석한 결과를 정리한 에너지 도표이다. 이 결과를 통해 아민 수용액에서 이산화탄소가 흡수되는 반응은 아민+CO2+아민의 모델로 계산하는 것이 활성화 에너지 측면에서 가장 적합하다고 제안했다. 또한 이 모델에서 반응물 아민과 염기 역할을 하는 아민을 서로 다른 분자로 설계하였을 때 활성화 에너지에서 차이가 생겨서, 아민 혼합물을 사용했을 때 CO2 흡수율이 높아지는 효과도 잘 보여준다는 것도 제시했다. 이와 같이 용액에서 일어나는 반응을 계산화학적 방법으로 연구하여 결과를 예측하는 것은 실제 반응을 관측하는 것에 비해 노력과 비용을 절감할 수 있다는 장점이 있다.
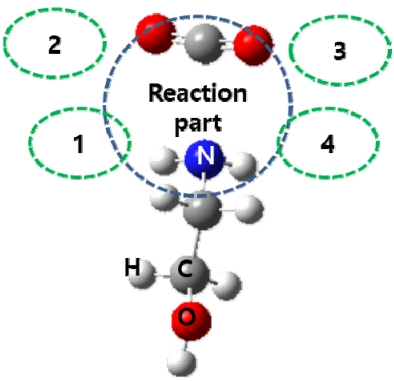
본 연구에서는 이전 연구 14) 에서 관찰한 반응이 일어나는 부분 주변에 아민과 물 분자를 추가하여 계산화학적으로 분석함으로써 그 결과를 이전 연구 결과와 비교하였다. 따라서 계산 모델은 MEA 수용액에 CO2가 흡수될 때 일어날 수 있는 모든 반응을 살펴보는 것이 아닌, 이전 연구의 모델에 주변 분자를 추가하는 방식으로 설계하였다. Fig. 2에는 이번 연구의 반응물 모델 설계 방식을 간단하게 나타냈고, Table 1에는 본 연구에서 설계한 14가지 모델에서 반응부 주위의 분자 배열을 정리했다. 아민은 가장 간단한 알카놀아민인 MEA를 사용하였고, MEA에 CO2가 결합을 형성하는 사이트 주변으로 최대 4개까지의 MEA 또는 물 분자를 배치하였으며, 1의 위치에는 반응에서 수소 원자 전달의 역할을 하는 분자를 놓았다. 이와 같은 방식의 설계로 이전 연구에서 사용한 [MEA+CO2], [MEA+CO2+H2O], [MEA+CO2+MEA] 이외에 [MEA+CO2+H2O+ MEA 또는 물 분자 1~3개]와 [MEA+CO2+ MEA+MEA 또는 물 분자 1~3개]를 추가하여 반응 모델을 크게 5가지 유형으로 구분하였으며, 세부적으로는 14가지 모델을 비교 분석하였다. 14가지 반응 모델의 구조를 모두 분석한 결과 각 유형으로 구분한 모델들에서 구조적인 특징이 크게 다르지 않아서 논문에서는 Table 1에서 ⓐ~ⓗ로 표시한 모델의 구조 계산결과를 제시하고 구조적인 분석을 서술하였다. CO2 흡수 공정에서 주로 사용하는 30 wt % MEA 수용액의 농도를 분자수의 비율을 따지면 MEA:H2O는 대략 1:8 정도가 되지만, 이러한 조건을 MD(molecular dynamics) 계산한 결과에 따라 제안된 반응 모델[23]과 용매 재생, 경제성 및 장치 부식방지 등을 고려하면 이번 연구의 계산 모델 중 ⓔ~ⓗ와 같은 분자 비율이 실제 공정을 비교적 잘 모사한 모델에 해당한다고 할 수 있다. 본 논문에서는 공정 조건 뿐만 아니라 다양한 분자 배열을 고려한 모델을 설계하여 반응 과정에서 주변 분자들의 영향을 비교하고자 한다.
3. 결과 및 고찰
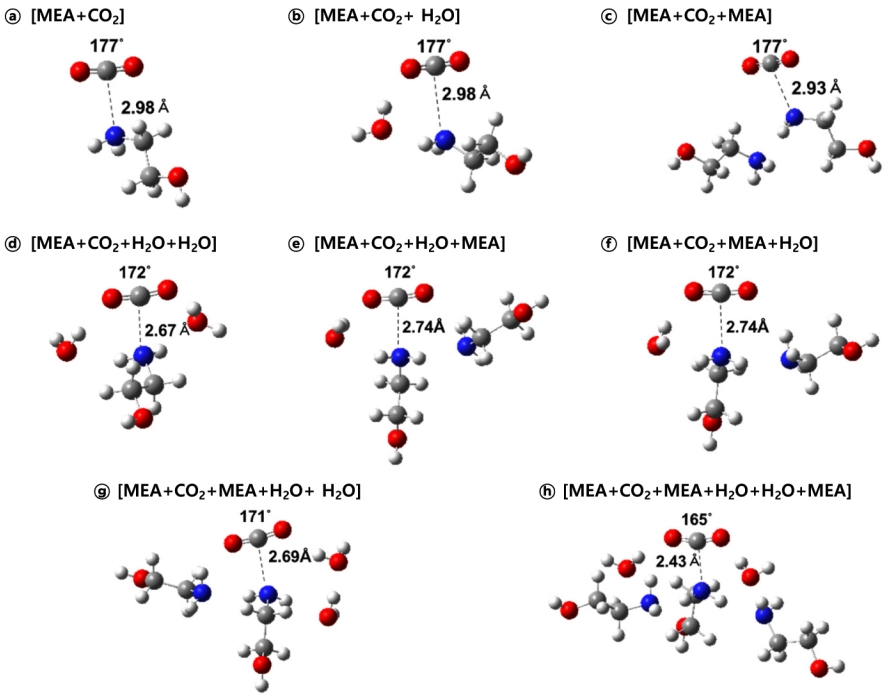
Fig. 3은 분석한 14가지 반응 모델 중 8가지의 반응물 복합체(reactant complex)를 나타낸 것으로, 반응물과 염기, 주변의 아민 또는 물 분자들을 Fig. 2와 같은 방식으로 배치하여 구조 최적화 계산을 한 결과이다. 그림은 여러 개 분자가 상호작용 하는 것을 고려하여 배치가 계산된 결과이기에 분자들이 따로 떨어져있는 것과는 다른 상태를 보여주므로 복합체(complex)라고 표현했다. 따라서 본 논문에서 제시되는 구조들은 모두 복합체 형태이다. ⓐ~ⓗ 모두 CO2의 탄소 원자는 반응물인 MEA의 질소 원자 중 비공유 전자쌍 쪽으로 향해있고, 이 두 원자들 간의 거리가 가까워짐에 따라 달라지는 C-O-C의 각도를 추가로 표시하여 비교했다. 반응물 이외에 추가한 MEA 또는 물 분자는 반응부(reaction part) 주변에서 반응물과 수소 결합을 잘 형성하는 위치로 배열되었고, ⓓ~ⓗ의 구조는 ⓐ, ⓑ, ⓒ에 비해 많아진 분자들간에 형성된 인력으로 인해 MEA의 질소 원자와 CO2의 탄소 원자 사이의 거리가 다소 가깝게 나타났다. CO2의 C-O-C의 각도도 ⓓ~ⓕ는 172o, ⓖ는 171o, ⓗ는 165o로 주변 분자와의 상호작용이 커질수록 점점 작아졌다. 이러한 구조는 CO2가 아민에 흡수되어 생성되는 카바민산, 양쪽성 이온 또는 카바메이트의 구조와 유사한 형태이다. 이런 변화는 실제 반응이 일어날 때 반응부 주변에 존재하는 많은 분자들의 영향으로 인해 분자간 상호작용이 강해져서 위에 제시한 세가지 메커니즘과 같은 CO2 흡수 반응이 잘 일어날 수 있는 형태로 반응물의 분자 구조나 배열이 변화할 수 있음 보여준다.
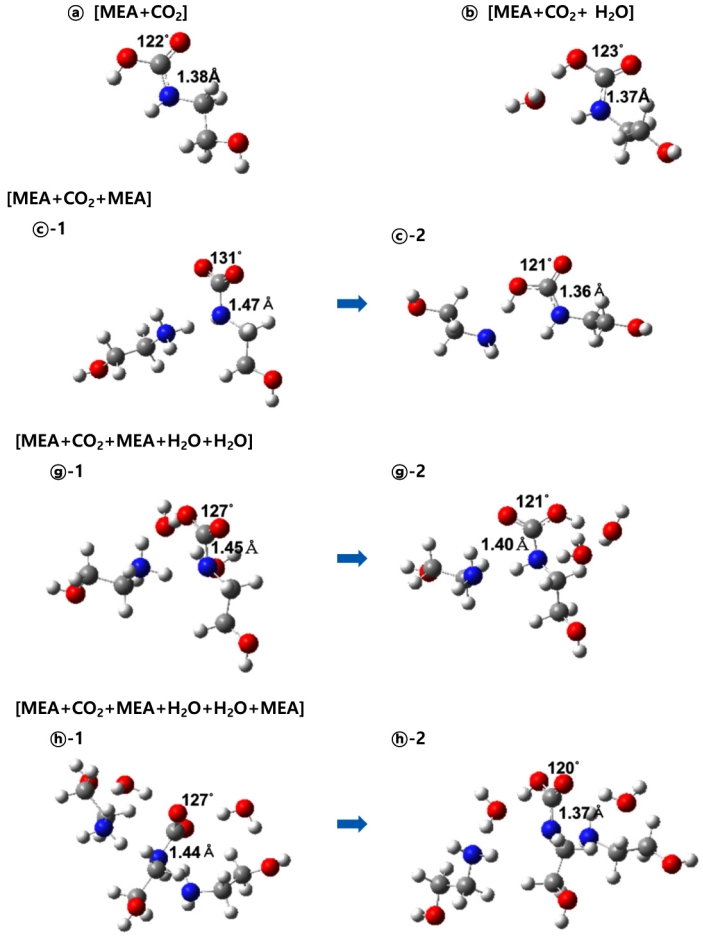
Fig. 4는 Fig. 3에서 살펴봤던 6가지 모델의 반응물로부터 얻어진 전이상태 구조이다. 이전 연구에서 ⓑ 모델은 두 개의 전이상태를 보이는 것으로 보고했는데, 그림에는 활성화 에너지가 큰 전이상태-2의 구조만을 나타냈다. 전이상태 구조들은 모두 반응물 구조에 비해 CO2가 MEA 쪽에 더 가까워져서 C-N 간의 거리는 약 1.5Å 정도이고, MEA의 아미노기의 수소 원자 한 개가 CO2(ⓐ)나 물(ⓑ, ⓓ, ⓔ) 또 다른 MEA (ⓒ, ⓕ, ⓖ, ⓗ) 쪽으로 이동하는 중간 상태의 구조임을 알 수 있다. 이러한 움직임은 그림에서 빨간색 점선으로 표시한 전이 상태 구조의 진동수 계산 결과, 한 개의 음의 진동수가 나타내는 원자의 이동에서 추가로 확인했다. 하지만 ⓑ, ⓓ, ⓔ는 다른 구조와 다르게 MEA의 아미노기의 수소 원자 한 개가 물 분자로 이동하는 것뿐만 아니라 물 분자의 수소 원자 한 개가 CO2로 이동하는 움직임이 동시에 일어났다. 이러한 차이로 인해 다른 구조들에 비해 ⓑ, ⓓ, ⓔ에서 CO2의 C-O-C의 각도는 약 127˚로 카바민산(120o)에 가장 가까운 구조를 보였다. 농도가 낮은 MEA 수용액에 CO2가 흡수될 때 카바민산이나 중탄산 이온이 생성된다는 연구결과를 고려할 때[6,24], ⓑ, ⓓ, ⓔ는 카바민산 생성과정을 잘 보여주는 전이상태 구조라 할 수 있다. 그림에 나타난 모델 외에도 물 분자가 MEA 아미노기의 수소 원자 전달의 역할을 하는 다른 모델에서는 모두 이와 유사한 전이상태 구조가 계산되었다. ⓒ, ⓕ, ⓖ, ⓗ와 더불어 또 다른 MEA 분자(반응부 주위에 위치한 분자)가 MEA(반응물) 아미노기의 수소 원자 전달의 역할을 하는 다른 모델(Table 1에서 III, V)에서 확인한 전이상태는 모두 ⓒ와 매우 유사하게 CO2의 C-O-C의 각도가 130 o 정도였다. 이전의 연구 결과와 비교하면 반응부 주변에 추가한 분자들이 새로운 결합을 형성하거나 수소 원자 전달 반응에 참여하는 것 같지는 않았다. 하지만 전이상태에서 반응부의 구조 변화에 따라 추가된 분자들의 배치가 꽤 많이 변하면서 분자간 인력이 강하게 작용하는 형태의 배열로 그 위치가 바뀌어 전반적인 구조가 다소 복잡해졌다.
Fig. 5는 5가지 모델에서의 생성물 구조를 나타낸 것이다. Fig. 4에서 확인한 전이상태 구조에서 추가로 넣어 준 물 또는 MEA 분자로 인해 새로운 결합이 형성되지 않는 것을 확인했기 때문에 생성물의 구조는 ⓐ, ⓑ, ⓒ, ⓖ, ⓗ의 결과만 나타냈다. Fig. 3, 4와 연결하여 서술하기 위해 각각의 구조에서 기호 표시는 위와 동일하게 했다. 전이상태 구조를 통해 생성물로 얻어진 ⓒ-1은 추가적인 구조 최적화 계산 결과 ⓒ-2와 같이 카바민산의 구조로 안정화되었으며, ⓖ와 ⓗ도 동일한 결과를 보였다. 이전 연구에서는 ⓒ 모델의 생성물이 카바메이트라고 보고했었지만[14], 추가적인 구조 최적화 계산을 수행한 결과 카바메이트 형태에서 카바민산으로 안정화된다는 것을 알 수 있었고, 본 연구에서는 이를 생성물로 했다. 카바메이트, 카바민산, 양쪽성 이온은 서론에서 제시한 반응 메커니즘에서 서술한 바와 같이 알카놀아민의 질소에 결합한 원자를 기준으로 구분하였고, Fig. 5에 나타낸 모델뿐 만 아니라 나머지 모델의 결과에서도 생성물이 모두 카바민산과 유사한 형태의 구조를 보였다. ⓐ, ⓑ, ⓒ-2, ⓖ-2, ⓗ-2에서 CO2의 탄소와 MEA의 질소 원자 사이의 거리는 약 1.4Å, O-C-O의 각도는 약 120o, C-N-CO(OH)는 모두 한 평면에 위치하는 구조를 확인할 수 있는데, 이는 카바민산의 구조와 매우 유사하다[25,26]. 14가지 모델에서 계산된 모든 생성물의 구조는 카바민산과 유사한 구조로 확인되었기 때문에 서론에서 제안한 세가지 유형의 반응 메커니즘 중 (1)의 카바민산을 중간체로 하는 반응을 잘 보여준다고 할 수 있다. Fig. 4의 전이상태를 거쳐서 Fig. 5와 같은 구조를 가진 카바민산 생성 반응이 잘 일어나면 최종 생성물인 카바메이트가 생성될 확률이 높아질 것이다. 게다가 (1)과 (2)의 반응 메커니즘 중 (1)이 느린 반응(반응속도결정단계, rate-determining step) 6) 에 해당하니 카바민산 생성 반응이 잘 일어나는지가 최종 생성물이 생성되는데 중요한 역할을 한다고 할 수 있다. 본 연구의 계산 결과에 따르면 중간체인 카바민산을 생성하는 (1)의 반응에서 반응물인 CO2와 MEA 이외에 반응부 주위에 위치한 물이나 MEA 분자는 염기가 아니라 수소 원자 전달 반응의 촉매의 역할을 했다고 볼 수 있다. 이는 Fig. 4의 전이상태 구조에서 반응에 참여한 주변 분자(MEA 또는 물)가 Fig. 5의 생성물 구조에서 수소 원자를 추가로 받지 않고 반응물에서와 같은 구조(MEA 또는 물)를 보이는 것을 통해 확인할 수 있다. 이는 주변 분자를 추가로 넣어 준 II~V의 유형의 13가지 모델에서 모두 같은 결과를 보였다. 이들 분자의 촉매 역할에 따른 활성화 에너지 값의 비교는 아래에서 논의하겠다.
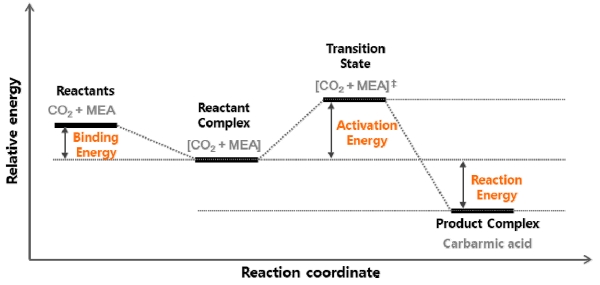
Fig. 6은 계산을 통해 얻어진 구조들의 에너지 차이를 비교하기 위해 바인딩 에너지(binding energy), 활성화 에너지(activation energy)와 반응 에너지(reaction energy)를 정의하는 에너지 도표이다. 앞에서 설명한 것과 같이 반응물 구조에 해당하는 분자들의 단순한 나열을 반응물(reactants), 이들의 배치를 구조 최적화 계산한 결과를 반응물 복합체(reactant complex)로 하면 도표에서와 같이 분자간 인력이 고려된 복합체의 에너지가 더 낮아진다. 반응물과 반응물 복합체의 에너지 차이를 바인딩 에너지라 정의하고, 계산된 바인딩 에너지가 음의 값이면 반응물보다 반응물 복합체의 에너지가 낮아진 것을 의미한다. 도표에 표시된 것과 같이 활성화 에너지는 반응물 복합체와 전이상태의 에너지 차이로, 반응 에너지는 반응물 복합체와 생성물 복합체의 에너지 차이로 정의했다. 이렇게 계산된 에너지 값을 Table 2에 나열하였다. 표에는 계산 모델에서 서술한 5가지 유형의 14가지 모델에 대한 계산 결과를 모두 나타냈으며, Fig. 3~5에 나타낸 모델명은 그림과 동일하게 ⓐ~ⓗ로 표시했고, 나머지는 기호 표시 없이 구성 분자들만 나열했다. 그리고 모델명에 나열된 분자들 중 반응에서 수소 원자 전달의 역할을 한 분자를 CO2 다음에, 파란색으로 표시했다. 모든 모델에서 반응물 각 분자들의 에너지를 더한 값에 비해 반응물 복합체의 에너지가 더 낮아졌고, 반응부 주위에 추가한 분자의 수가 많아질수록 바인딩 에너지의 절대값이 커지는 것을 알 수 있다. 앞에서 확인한 반응물 복합체의 구조와 바인딩 에너지 값을 통해 실제 반응에서 반응물 주변에 존재할 많은 분자들은 구조의 안정성을 높이는 역할 뿐만 아니라, 반응이 일어나기에 용이한 방향으로 분자들이 배열되는데 도움을 줄 수 있다는 것 알 수 있다. 수소 원자 전달의 촉매가 고려되지 않은 I 유형의 활성화 에너지는 37.60 kcal로 14가지 모델 중 가장 높아 실제로 이러한 형태의 반응은 일어나기 어려울 것이다. 물 분자가 수소 원자 전달의 역할을 하는 II와 IV 유형의 활성화 에너지는 14.98~18.52 kcal이고, 해당 에너지는 주로 수소 원자 한 개가 MEA의 아미노기에서 물 분자를 거쳐서 이산화탄소의 산소 원자 쪽으로 전달되는 과정에서 쓰였다. MEA 분자가 수소 원자 전달의 역할을 하는 III과 V 유형의 활성화 에너지는 1.60~5.53 kcal이고, 해당 에너지는 주로 수소원자 한 개가 MEA의 아미노기에서 또 다른 MEA의 아미노기 쪽으로 전달되는 과정에서 쓰였다. 물이나 MEA가 촉매의 역할을 하는 모델의 활성화 에너지는 I 유형에 비해 낮았지만, II와 IV보다는 III과 V의 유형에서 활성화 에너지 값이 훨씬 낮다. 활성화 에너지는 반응물 주변 분자들의 수와 무관하게 전이상태에서의 수소 전달 과정의 차이로 인해 II-IV와 III-V에서 차이가 났다. 반응 유형에 따라 큰 차이를 보이는 활성화 에너지 결과를 통해 MEA가 수소 원자 전달의 역할을 하는 반응이 실제로 일어날 가능성이 가장 높을 것임을 확인할 수 있었다. 반응 에너지는 -7.66~4.80 kcal로 반응이 발열반응일수도 흡열반응일 수도 있는 것으로 계산되었다. 표의 결과를 전체적으로 보면 활성화 에너지는 반응 모델의 유형에 따라 값의 차이가 분명하지만, 반응 에너지는 그렇지 않다. 실제 공정에서 MEA 수용액에서 CO2 흡수 반응은 발열반응이지만, 카바민산은 반응 중간체이기 때문에 표의 반응 에너지로는 이를 제안할 수는 없다. 하지만 실제 공정과 유사한 분자수의 비율과 활성화 에너지를 고려하면 실제 반응을 가장 잘 보여주는 모델은 ⓕ, ⓖ, ⓗ라고 할 수 있다. MEA와 CO2의 반응에서 주변에 추가로 배치한 분자들의 영향을 보면, 주변의 분자들 중 촉매의 역할을 하는 분자의 유무에 따라 전이상태 구조와 활성화 에너지 값의 차이가 크지만, 촉매 이외의 분자들의 영향은 크지 않은 것으로 보인다.
4. 결 론
본 연구에서는 MEA 수용액에 CO2가 흡수되는 과정에서 반응에 참여하는 분자 이외에 반응부 주변에 존재할 많은 분자들의 영향을 계산화학적으로 분석하였다. 반응부 주변에 추가한 MEA 또는 물 분자의 수를 1~4개로 달리하여 5가지 유형의 14가지 반응 모델의 구조와 에너지를 비교하였다. 반응물인 MEA와 CO2 이외에 추가로 MEA나 물 분자 한 개씩까지만 추가하여 반응 모델을 설계했던 이전 연구의 결과와 비교하면 반응물 복합체의 구조에서 가장 큰 차이가 있었다. 반응부 주변에 추가한 분자들의 수가 증가함에 따라 반응물과 수소결합을 형성하면서 반응물 복합체가 안정화되어 바인딩 에너지의 절대값이 증가했다. 또한 CO2 흡수 반응에 용이한 방향으로 반응물 복합체의 구조가 바뀌는 것을 확인할 수 있었다. 실제 반응이 일어날 때에는 이보다 더 많은 MEA와 물 분자들이 반응부 주변에 있을 것이므로 이 분자들에 의한 영향이 더욱 커질 수 있다. 이렇게 안정화된 반응물 복합체는 전이상태에서 수소 원자 전달 과정을 거쳐 생성물(전체적으로는 반응 중간체)인 카바민산으로 바뀌게 된다. 전이상태나 생성물에서도 반응부가 주변에 추가로 넣어 준 분자들과 강한 인력을 형성하고 있긴 하지만, 반응 과정은 변하지 않았다. 특히, 추가된 분자들과 무관하게 활성화 에너지는 수소 원자 전달의 역할을 MEA가 하는지, 물이 하는지에 따라 달라졌다. 이 차이는 수소 원자 전달 과정이 두 가지 유형에서 서로 다르기 때문이다. 수소 원자 전달을 MEA가 하는 유형(III, V)에서는 아미노기의 수소 원자 한 개가 MEA 쪽으로 이동하지만, 물이 하는 유형(II, IV)에서는 아미노기의 수소 원자 한 개가 물 쪽으로 이동함과 동시에 물의 수소 원자 한 개가 이산화탄소 쪽으로 이동한다. 이러한 차이로 인해 활성화 에너지는 전자에서 1.60~5.53 kcal, 후자에서 14.98~18.52 kcal으로 계산되었다. 활성화 에너지를 기준으로 이 반응을 분석해보면 CO2를 흡수하는 실제 반응에서 수소 원자 전달의 역할은 MEA가 하는 방식으로 일어날 것이라 해석할 수 있고, 이는 이전 연구들을 통해서도 잘 알려진 결과이다[7,13,14,27,28].
계산화학적으로 반응의 메커니즘을 연구할 때, 반응물 이외에 주변에 존재할 무수히 많은 분자들의 영향을 어떻게 고려해야 적당한가에 대한 문제는 항상 대두되기 마련이다. 따라서 본 연구는 이러한 의문을 확인하기 위해 다양한 계산 모델을 설계하여 그 결과를 비교 분석하였다. 이번 연구를 통해서 반응부 주위에 배치하는 분자의 수를 늘리는 것은 반응 에너지와 각 분자들의 상호작용에 의한 배치에는 많은 영향을 주었지만, 전이 상태의 구조에서 변화가 일어나는 방식과 활성화 에너지에는 그 영향이 크지 않다는 것을 확인할 수 있었다. 모든 반응에서 이와 같은 경향성이 유효하지 않을 수는 있지만, 본 연구와 유사한 유형의 반응에 대한 연구에서는 충분히 고려할 만한 결과일 것이다.