DNA 추출법에 따른 혐기소화조 미생물 군집 분석 결과 비교
Comparison of DNA Extraction Methods for Metagenomic Sequencing in Anaerobic Digestion
Article information

Abstract
목적
DNA 추출 방법에 따른 혐기소화조 시료의 미생물 군집 분석 결과를 비교한다.
방법
실규모 중온 혐기소화조 시료를 동일 볼륨으로 소분하였다. DNA 추출법으로는 국내에서 많이 사용하는 제품 중 미국 MP Biomedicals사의 FastDNA spin kit for soil, 한국 GeneAll사의 Exgene stool DNA mini kit, 그리고 한국 Bioneer사의 AccuPrep genomic DNA extraction kit의 세 종류를 사용하였다. 박테리아의 경우 V3-4, 고세균의 경우 V4-5 region에 해당하는 16S rDNA amplicon을 제작한 뒤, 일루미나 iSeq 100 장비로 염기 서열을 분석하였다. 미생물 군집 분석을 위해서 quality filter를 거친 유효한 read만을 얻은 뒤, OTU clustering을 하여 RDP classifier를 이용해 계통 분석(taxonomic assignment)하였다.
결과 및 토의
Weighted UniFrac distance로 군집 결과를 시각화한 결과, 박테리아와 고세균 모두 사용한 세 가지 kit에 따라 군집 구조 차이를 보였으며, 특정 군집들은 DNA 추출 방법에 따라 존재 비율의 차이가 크게 나타났다. 예를 들어, soil kit은 Proteobacteria 문에 대한 추출 효율이 다른 추출법에 비해 낮고, Thermotogae에 대해서는 높은 것으로 나타났으며, stool kit은 이와 반대의 경향성을 보였다. 고세균의 경우, genus 중 Methanomethylovorans와 Methanosarcina가 DNA 추출법의 영향을 특히 많이 받는 것으로 나타났다.
결론
미생물 군집 결과는 DNA 추출 방법에 영향을 받았다. 동일 선상의 실험에서 미생물 군집을 비교하려면 반드시 동일한 DNA 추출 방법을 사용해야 한다는 것을 확인하였다.
Trans Abstract
Objectives
This study was performed to compare three commercial kits for the extraction of genomic DNA from anaerobic digestate for subsequent iSeq 100 sequencing and microbial community analysis.
Methods
A full-scale mesophilic biogas plant was sampled, and made into aliquots of identical volumes to extract DNA using three commercial kits: FastDNA spin kit for soil (MP Biomedicals, USA), Exgene stool DNA mini (GeneAll, South Korea) and AccuPrep genomic DNA extraction kit (Bioneer, South Korea). To analyze the microbial communities in the purified DNA, 16S rDNA amplicon sequencing (V3-4 region for bacteria and V4-5 region for archaea) was performed on the Illumina iSeq 100 platform. Quality filtered sequence reads were OTU-clustered for taxonomic assignment conducted using the RDP classifier on-line.
Results and Discussion
The microbial community structure visualized on the NMDS plot using the weighted UniFrac distance revealed that both bacteria and archaeal communities have phylogenetic differences depending on the DNA extraction kit used. In addition, the abundance of certain microbial populations was significantly different among the DNA extraction methods. For example, Proteobacteria was the least abundant using the soil kit, while this phylum was the most abundant when the stool kit was used. However, in the case of Thermotogae, this tendency was vice versa. The abundance of archaeal genera Methanomethylovorans and Methanosarcina was also affected by DNA extraction methods.
Conclusions
The microbial populations analyzed by 16S based sequencing were affected by DNA extraction methods. To compare microbial community changes in the identical set of research, one DNA extraction method should be chosen and used consistently for the whole experiment.
1. 서 론
오늘날 차세대염기서열분석(Next-generation sequencing, NGS) 기술이 보편화됨에 따라 개별 실험실 수준에서도 관련 장비를 구비하여 다양한 환경 시료에 존재하는 미생물의 군집 구조를 신속하게 분석할 수 있게 되었다[1,2]. 환경 시료와 같이 reference genome을 특정할 수 없고, 보고된 전체 genome database를 대상으로 유사 서열을 탐색하는 경우를 메타지놈(metagenome) 분석이라고 일컫는다. 미생물의 메타지놈 분석을 위해서는 shotgun 방법을 쓸 수도 있으나, 보다 경제적이고 간편한 방법으로서 16S rRNA 유전자를 대상으로 하는 amplicon sequencing 방법이 널리 쓰인다. 이것은 통상 박테리아 또는 고세균에 보존된 서열(conserved sequence)을 프라이머(primer)로 하고, 그 사이의 분류학적 다양성이 큰 부분(hypervariable region)을 PCR 반응으로 증폭시킨 뒤, 해당 산물(amplicon)의 염기 서열을 읽는 방식에 해당된다[1].
메타지놈 분석은 여러 단계의 실험과 분석을 거쳐 진행될 뿐 아니라, 얼마나 실제 군집 구조에 가깝게 분석되었는지 실험의 정확성을 기준할 만한 방법이 없기 때문에, 비교하고자 하는 일련의 시료를 최대한 동일한 방법으로 분석해야 한다. 샘플링 방법뿐만 아니라, DNA 추출[3,4], 시퀀싱 플랫폼[5], 그리고 분석 알고리즘[6,7] 등 과정에서 이질적인 방법을 사용할 경우 결과의 일관성을 보장할 수 없기 때문이다.
특히, 미생물은 균주(strain)에 따라 다양한 세포막과 세포 외벽의 구조를 가지므로, 생물학적 다양성이 높은 환경 샘플의 경우 세포 용해 및 DNA 추출 방법에 따라 분석된 군집 구조가 달리 나타나게 된다[8]. 세포를 용해하는 방법으로는 bead beating, dry-thaw의 반복 등과 같은 물리적 파쇄(mechanical disruption) 단계를 거칠 수도 있고, proteinase K나 라이소자임(lysozyme)과 같은 생물학적 효소 처리를 할 수도 있으며, 화학적 방법으로서 이온성 혹은 비이온성의 다양한 계면 활성제를 선택적으로 사용한다[9]. 균주에 따라 세포 용해방법에 반응하는 정도가 다르며, 이 외에도 DNA 추출 수율은 DNA 분자의 크기, 염기 조성, topology 및 단백질과의 결합 상태에 따라 영향을 받으므로[4], 실험자는 실험에 사용할 DNA 추출 방법이 어떤 특성을 가지고 있으며 대상 시료에 적합한지 확인할 필요가 있다.
혐기소화는 가축분뇨, 하수슬러지, 음식물류폐기물 등 유기성 폐기물을 기질로 사용하여 바이오가스를 생산하는 친환경적인 폐기물 자원화 공법 중 하나이다. 미생물의 생화학적 활성이 공정의 주요 반응 인자이므로, 공정 최적화 및 운영 안정성 향상을 위해 혐기소화 미생물 군집 구조를 규명하는 연구가 널리 수행되고 있다[10,11].
본 연구에서는 혐기소화조 시료에 존재하는 미생물을 16S amplicon sequencing으로 분석할 때, DNA 추출법에 따라 군집 결과가 어떻게 달라지는지 비교하였다. 세포 용해에 있어 물리적 파쇄를 이용하며 전 세계적으로 널리 쓰이고 있는 soil kit, 장내 미생물 판별을 위해 쓰이는 stool kit, 효소 처리를 이용하며 다양한 샘플에 사용될 수 있는 범용 genomic DNA (gDNA) kit을 실험에 사용하였다. 각 방법으로 추출한 DNA에서 박테리아 및 고세균에 대한 16S amplicon sequencing을 수행한 뒤, 군집 결과를 phylum(박테리아), genus(고세균) 단계 및 weighted UniFrac으로 계산하여 비교하였다.
2. 실험방법
2.1. DNA 추출
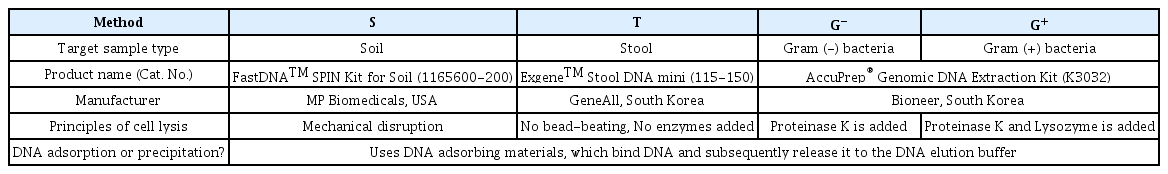
울산광역시 용연하수처리장의 중온 혐기소화조(하수슬러지와 음식물류폐기물을 병합 소화) 시료를 샘플링하여 850 µm 체로 거른 뒤, 질소로 가스 치환하여 혐기 상태로 35℃에서 추가 기질의 투입 없이 바이오가스가 더 이상 발생하지 않을 때까지 배양하였다(최종 17.2 g VS/L). 이 배양액을 동일 볼륨(40 µL)으로 마이크로 튜브에 소분하였고, 10,000×g에서 5분 원심분리하여 상등액을 제거하였다. 이후 세포 용해(cell lysis) 단계부터 각 추출법에 따라 2회 반복으로 진행하였다. DNA 추출법으로는 국내에서 많이 사용하는 제품 중 세 종류의 kit를 선택하여 총 네 가지 방법으로 각 2회 반복하여 추출하였다. 미국 MP Biomedicals사의 FastDNA spin kit for soil(방법 S), 한국 GeneAll사의 Exgene stool DNA mini kit(방법 T), 그리고 한국 Bioneer사의 세포 종류에 따라 세부 옵션을 선택하여 사용하는 AccuPrep genomic DNA extraction kit(그람 음성 세균용: 방법 G+, 그람 양성 세균용: 방법 G-)을 사용하였다. 실험 과정은 Fig. 1에 도표로 나타내었으며, 각 추출법에 대한 소개와 원리를 Table 1에 정리하였다.

The experimental workflow.
DNA extraction methods used in this experiment.
혐기소화조 시료의 특성에 맞추어 세포 용해 단계가 다소 수정된 부분이 있으며, 보다 구체적으로 기술하면 다음과 같다. 방법 S의 경우, 상등액을 제거한 pellet에 전용 lysis buffer (sodium phosphate buffer with MT)를 넣어 resuspension한 뒤, 실리카 및 세라믹 재질의 입자가 담긴 2 mL microtube에 넣고 mini-beadbeater-1 (Biospec products, USA)을 이용해 4200 rpm 속도로 40초간 파쇄하였다. 방법 T의 경우 stool 시료가 아니므로 homogenization 단계를 생략하고, pellet에 lysis buffer (FL)를 넣고 이후는 kit에 제시된 프로토콜대로 진행하였다. 방법 G-는 주어진 lysis buffer (TL)에 proteinase K를 첨가하여 60℃에서 1시간 처리하였고, 방법 G+는 별도로 제시된 lysis buffer (20 mM Tris-HCl, pH 8.0; 2 mM EDTA; and 1.2% Triton X-100)에 라이소자임을 넣고 37℃ 30분 처리한 뒤, 또다른 lysis buffer (GB)를 넣고 proteinase K 첨가하여 60℃ 30분 반응시켰다. 방법 G-와 G+의 경우 세포 용해가 끝난 반응액을 12,000 ×g에서 5분간 원심분리하여 상등액만 분리하여 다음 단계로 진행하였다. DNA 용출 단계에서는 각 kit에 포함된 elution buffer를 동량(100 µL) 넣어 진행하였으며, 추출된 DNA의 농도는 Qubit 4 fluorometer (ThermoFisher)로 측정하였다.
2.2. 16S metagenomic sequencing
추출한 DNA에 존재하는 미생물 군집을 알아내기 위해 Illumina iSeq 100 장비를 이용하여 제시된 프로토콜대로 실험하였다. 먼저, 정제된 DNA를 주형으로 하여 amplicon PCR을 수행하였다. 사용한 PCR 프라이머는 박테리아 또는 고세균(archaea) 특이적인 16S rDNA 서열과 함께 Illumina overhang adapter 서열이 추가된 올리고머를 사용하였다. 박테리아 특이적인 증폭을 위해서는 518F(5’-CCAGCAGCCGCGGTAATACG-3’) 및 805R(5’-GACTACCAGGGTATCTAATCC-3’) 서열을 포함하였고, 고세균 군집 규명을 위해서는 787F(5’-ATTAGATACCCSBGTAGTCC-3’)와 1059R(5’-GCCATGCACCWCCTCT-3’) 서열을 포함하였다. PCR 산물을 정제한 뒤 index PCR을 수행하였고, 45 pM로 library pooling 및 PhiX sequencing control과 혼합하여 염기서열 분석을 진행하였다.
2.3. 미생물 군집 분석
얻어진 paired-end read 서열을 merge 및 quality-filter (maximum expected errors=1; minimum length=200 및 chimera check)한 뒤, VSEARCH 알고리즘을 이용하여 sequence-identity cutoff를 97%로 지정하여 OUT (operational taxonomic unit) clustering하였다. 그 후 RDP classifier (https://rdp.cme.msu.edu/classifier/)를 통해 박테리아의 경우 4만 개 이상, 고세균의 경우 3만 개 이상의 유효한 서열(valid reads)을 얻어 계통학적으로 분류(taxonomic assignment)하였다.
모든 실험군에서 0.1% 이하로 존재하는 군집일 경우 minor population으로 분류하여 합산하였고, RDP classifier에서 confidence score가 80%보다 작은 OTU는 unclassified로 지정하여 합산하였다.
3. 결과 및 고찰
3.1. 추출된 DNA의 농도 측정
전술된 네 가지 방법으로 DNA를 분리 정제한 뒤 농도를 측정하였고, A260/A280 비는 모두 1.8 이상으로 도출되었다(Table 2). 방법 S의 농도를 100%로 하여 상호 비교한 결과, 정제된 DNA의 농도는 S>G->G+>T 순으로 나타났다. 방법 S로 추출된 DNA의 양과 비교하였을 때 T는 31%에 그쳤고, G+는 50%, G-는 63%로 나타났다.

The concentration of eluted DNA using different extraction methods.
방법 S는 흙에 존재하는 gDNA의 분리뿐 아니라, 하수슬러지 및 혐기소화조 샘플에서 미생물 DNA를 추출하는 데 널리 사용되는 방법이다. 별도의 enzyme 처리를 하지 않는다는 장점이 있으나, bead beating과 같은 물리적 파쇄를 이용한 방법은 DNA의 조각남(fragmentation) 현상이 다른 추출 방법에 비해 높다는 단점이 있다[12]. 따라서 DNA 구조가 최대한 보존되고 손상되지 않는 추출법을 원한다면 이 방법은 적절하지 않을 수 있다.
방법 T로 추출된 DNA의 양은 방법 S로 얻은 DNA 양과 비교해 31% 수준이었다. 혐기소화 공법은 동물의 장내 환경과 다소 유사하고[13] 투입 기질로 음식물류를 사용하는 경우가 많기 때문에[14,15] stool kit으로 혐기소화 미생물의 DNA를 추출하는 것이 적합할 수 있을 것이라고 추론하였으나, 해당 제품으로는 추출된 DNA의 농도가 가장 낮았다.
방법 G-는 그람 음성균에 특화된 방법으로서 proteinase K를 60℃에서 한 시간 처리하였다. 방법 G+는 그람 양성균의 세포벽 구성 물질인 펩티도글리칸(peptidoglycan)을 분해하기 위해 라이소자임을 37℃에서 30분 처리하고, 이어서 proteinase K를 30분 처리하였다. 방법 G+를 통해서도 그람 음성균의 세포 용해가 일어날 수 있음에도 불구하고 G-가 G+보다 DNA 추출 효율이 높은 이유는 고온(60℃)에서의 처리 시간이 G+가 더 길고, lysis buffer 조성(제품 설명에 명시되어 있지 않음)이 다르기 때문일 것으로 생각된다.
3.2. DNA 추출법에 따른 박테리아 군집 비교
박테리아 문(phylum)의 존재 비율을 Fig. 2에 나타내었다. 모든 샘플에서의 존재 비율이 0.1% 이하일 경우에는 minor population으로 분류하였다. 또한, 추출법에 따라 각 군집의 존재 비율이 얼마나 크게 영향을 받는지 알아보기 위해 각 군집의 CV (coefficient of variation)를 계산하였는데, CV는 각 추출법(S, T, G-, G+)에 해당하는 군집 비율의 평균값을 구한 뒤, 이들 네 개 값의 평균에 대한 표준편차의 비율로 정의하였다. 이것은 고세균의 속(genus) 분석에서도 동등하게 적용하였다.

The relative abundance of bacterial populations at phylum level.
점유율 10% 이상의 군집은 모든 시료에서 Thermotogae, Bacteroidetes, Firmicutes, Proteobacteria였다. 이 중 Thermotogae는 방법 S를 사용하였을 때 가장 높은 비율로 추출되었으며(33.17%), 방법 T를 사용하였을 때 가장 적게 추출되었다(22.01%). 한편, Proteobacteria는 앞서 언급한 우점 군집 중에서 가장 큰 CV를 나타냄으로써 추출법에 가장 영향을 크게 받는 phylum으로 나타났다. 방법 S를 사용했을 때에는 네 번째로 우점했으나(13.72%), 다른 추출 방법을 사용했을 때는 모두 상위 두 번째 우점 군집으로 나타났고, T 추출법을 사용했을 때 존재 비율이 가장 높았다(21.17%).
Phylum 단계에서의 박테리아 군집 차이를 시각화하고자 Bray-Curtis distance를 이용해 non-metric multidimensional scaling (NMDS) 도표를 나타내었다(Fig. 3(A)). 그 결과, 방법 S와 T 서로 떨어져 위치하고, 그 사이에 추출법 G+으로 얻은 군집 결과가 S와 G-에 가깝게 위치하는 양상을 보였다. 즉, 라이소자임을 투입한 방법 G+는 stool kit보다는 soil kit과 근접한 phylum 구조를 가진다는 것을 확인하였다.

NMDS plot of bacterial populations based on the (A) Bray-Curtis distance at the phylum level and (B) weighted UniFrac distance.
한편, OTU 간의 계통학적 거리(phylogenetic distance)와 상대 비율을 함께 고려하여 군집 구조의 차이를 시각화하기 위해 weighted UniFrac distance(α = 0.5)를 계산하여 박테리아 군집을 NMDS plot으로 나타내었다(Fig. 3(B)). 그 결과, G-와 G+가 같은 위치에 겹쳐져 총 세 개의 클러스터로 나타났다. 즉, 동일한 추출 kit을 사용하는 방법 G-와 G+는 phylum level만 고려했을 경우에는 라이소자임 투입 여부에 따라 군집 구조가 약간 차이가 있는 것으로 나타났으나(Fig. 3(A)), OTU에 대한 전체적인 계통학적 분류를 고려했을 시에는 매우 유사한 군집 구조를 가지는 것으로 나타났다. 세 개의 클러스터는 어느 한쪽에 치우치지 않았으므로 비교적 균등한 군집 차이를 보인다는 것을 확인하였다.
또한 각 추출법에 따른 군집 구조의 특징을 풍부도(richness), 다양성(diversity), 균등도(evenness) 방면에서 살펴보았다(Fig. 4). Rarefied richness는 동일 read 수를 가정했을 때 발견되는 OTU의 개수를 말하며, evenness는 OTU들이 얼마나 균등한 점유율(abundance)을 갖는지 나타내는 값이다. Shannon 및 Simpson index는 diversity의 지표로서 OTU의 richness와 evenness를 함께 고려하며, 두 값이 커질수록 다양성은 증가하게 된다.

Diversity indices of bacterial populations: rarefied richness, Shannon index, Simpson index and Pielou’s evenness index. Groups with a shared letter indicate no significant difference at p < 0.05 from the one-way ANOVA analysis (Duncan’s multiple range test).
방법 S와 G-을 사용하였을 때에는 풍부도, 다양성, 균등도의 값들이 모두 낮았고, 방법 T를 사용하였을 때는 모두 높게 나타났다. 방법 G+의 경우 rarefied richness와 Shannon index에서는 S와 T의 중간값을, Simpson index와 Pielou’s evenness index에서는 S와 비슷한 값을 보였다. 다양성과 균등도 지표들은 DNA 추출 농도(Table 1)와 대체로 역행하는 경향을 나타내었다. 즉, 방법 S와 같이 DNA 추출 농도가 높았던 경우가 상대적으로 낮은 다양성을 보였고, 방법 T와 같이 DNA 추출 농도가 낮았던 경우가 더 높은 다양성을 보였다. 방법 S의 물리적 파쇄가 일부 미생물의 세포벽을 더 높은 효율로 용해하였기 때문일 수 있으며, 물리적/효소적 파쇄를 이용하지 않는 방법 T가 상대적으로 가장 균등한 세포 용해를 나타낸 것으로
3.3. DNA 추출법에 따른 고세균 군집 비교
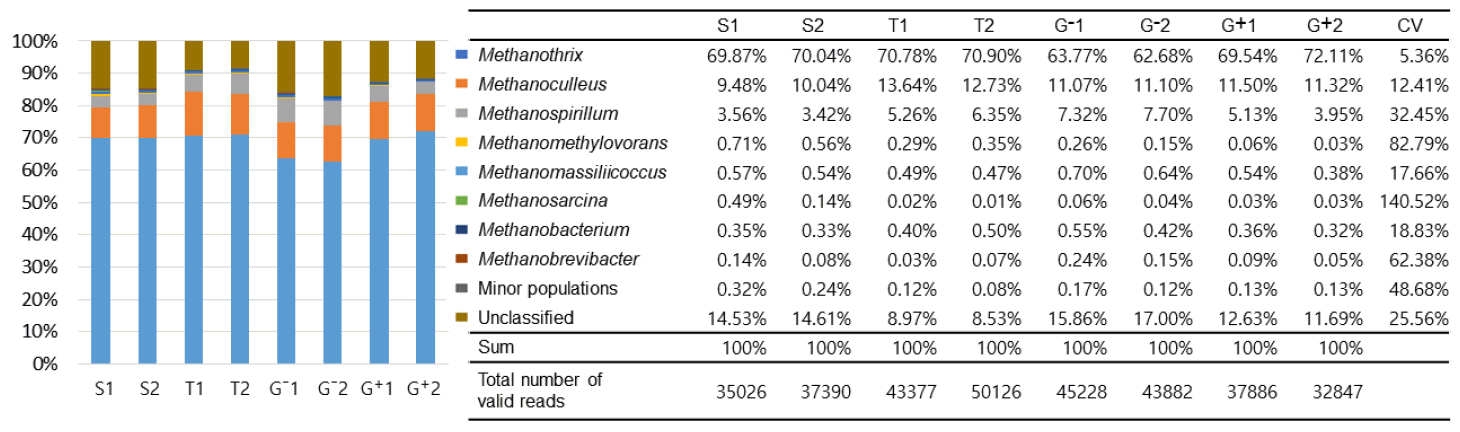
고세균의 genus level에서의 군집 결과를 Fig. 5에 나타내었다. 모든 실험군에서 Methanothrix의 존재 비율이 60~70%를 차지하여 가장 우점하였으며, 두 번째 우점군은 Methanoculleus였다.
The relative abundance of archaeal populations at genus level.
Methanomethylovorans는 방법 S에서 상대적으로 높은 비율을 보였고, G+에서 가장 낮았다(S, 0.64%; T, 0.32%; G-, 0.21%; G+, 0.05%). Methanosarcina 역시 S에서 높은 존재 비율을 나타냈고(0.56%), 그 외 추출법으로는 해당 군집의 비율이 현저히 낮았다(T, 0.02%; G-, 0.05%; G+, 0.03%). 따라서 이 두 군집의 존재 비율을 주의 깊게 살펴보아야 할 연구의 경우에는 S 추출법을 사용하는 것이 좀 더 민감하게 해당 군집 변화를 살펴보는 데 유리할 것이라고 판단된다.
Fig. 5의 결과를 Bray-Curtis distance로 NMDS plot에 나타낸 결과, S, T, G-는 비교적 균등한 차이를 보였고, G+(라이소자임 투입)는 G-(라이소자임 미투입)와 근접하기보다는 S와 T 중간에 자리하였다(Fig. 6(A)). 즉, 라이소자임 처리에 따라 고세균 군집이 달라지는 것을 확인하였다. Weighted UniFrac distance를 시각화한 결과, 마찬가지로 S, T, G-가 독자적인 군집 구조를 보였으나, 라이소자임을 투입할 경우(G+)에는 T와 근접한 결과를 얻었다(Fig. 6(B)). 즉, pseudopeptidoglycan을 가진 고세균 군집의 경우 라이소자임과의 반응 여부에 따라 genus level 및 weighted UniFrac distance에서 군집 구조가 다르게 나타나는 것을 확인하였다. 앞서 박테리아의 경우, 라이소자임과의 반응 여부에 따라 phylum level에서의 군집 차이는 다소 있었으나, weighted UniFrac에서는 미미하였다.

NMDS plot of archaeal populations based on the (A) Bray-Curtis distance at the genus level and (B) weighted UniFrac distance.
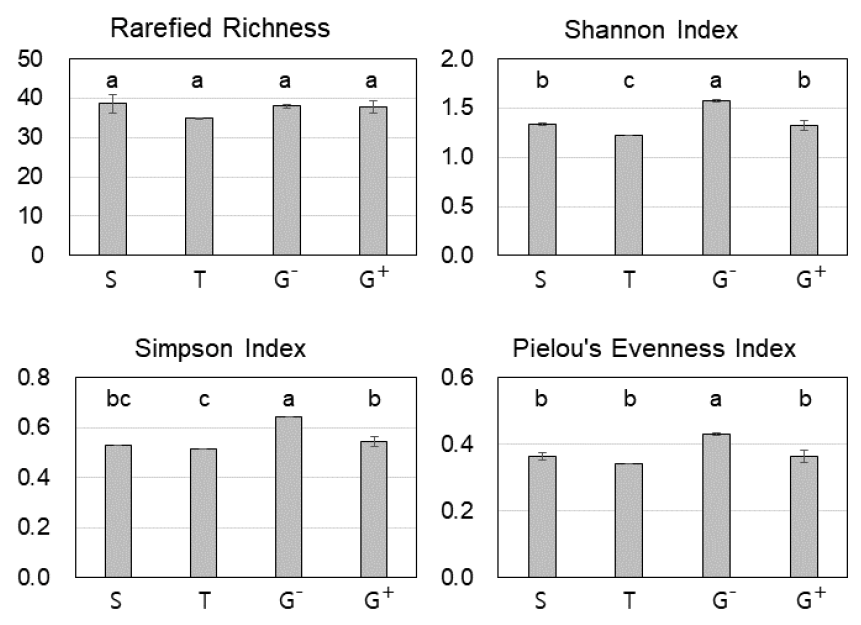
한편, 고세균의 풍부도(rarefied richness)는 모든 추출법에서 유사한 수치를 나타냈다(Fig. 7). 다양성(Shannon 및 Simpson index)과 균등도(Pielou’s evenness index)에서 방법 G-가 가장 높은 수치를 보였고, T가 가장 낮은 수치를 보였다. 즉, stool kit은 박테리아에 대해서는 다양성 수치가 가장 높게 나타났었으나, 고세균에 대해서는 우점 군집이 아닌 소수 군집을 다른 추출법보다 더 낮게 예측하는 경향성이 발견되었다. 방법 G+ 는 방법 G-와 동일 키트를 사용하며 라이소자임 처리를 추가한 것임에도 G-보다 고세균 다양성과 DNA 추출 농도(Table 1)가 더 작게 관측되었는데, 이러한 경향성은 박테리아 군집 다양성(Fig. 4)에서는 관측되지 않았다. 따라서 세포 용해 과정을 고도화하는 것이 무조건적으로 DNA 추출 효율이나 다양성을 높이지는 않으며, 서로 다른 세포벽 구조를 가지는 미생물 군집간에 그 효과가 다르게 적용된다고 해석된다.
Diversity indices of archaeal populations: rarefied richness, Shannon index, Simpson index and Pielou’s evenness index. Groups with a shared letter indicate no significant difference at p < 0.05 from the one-way ANOVA analysis (Duncan’s multiple range test).
4. 결 론
오늘날 NGS 장비의 보급이 널리 확대됨에 따라 실험실 차원에서 장비를 보유하여 일상적인 DNA 서열 분석이 가능해졌다. 그러나 메타지놈 연구는 환경 샘플과 같이 자연 그대로의 복잡한 생물 다양성을 가진 시료를 분석 대상으로 하며, 군집 분석 결과의 정확도를 측정할 수 있는 reference genome이 존재하지 않는다. 따라서 얻어진 NGS 결과가 얼마나 실제 군집 구조에 가까운지는 알 수 없다는 한계점이 존재한다.
본 연구에서는 DNA 추출법에 따른 미생물 군집 구조 결과 차이를 실험적으로 제시하였다. 그러나 이러한 경향성은 시료의 특성에 따라 달라질 수 있으므로 본 연구에서 통해 도출된 결과를 일반화하여 DNA 추출법의 우열을 판단하는 근거로 사용할 수는 없으며, 후속 연구에서는 real-time PCR을 이용한 박테리아와 고세균의 정량을 통해 DNA 추출법을 다각도에서 비교할 수 있을 것으로 판단된다. 이 연구의 결론으로서 분명한 것은 DNA 추출법에 따라 군집 구조의 편향성이 존재하며, 군집 구조의 변화 추이를 면밀히 살펴보는 일련의 실험에서는 동일한 DNA 추출법을 사용해야 한다는 것이다. 향후 다양한 환경 샘플을 분석하거나 배양된 미생물을 인위적으로 배합하여 DNA 추출법에 따른 군집 구조의 편향성을 조사한다면, 환경생물 관련 연구에서 true diversity를 관측하는데 도움이 될 수 있을 것이다.
Acknowledgements
본 연구는 2020년도 경남과학기술대학교 교원 연구 활성화 지원 사업의 예산 지원으로 수행되었습니다.
References
Notes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.